Introduction to Visualization of Protein Structures - May 2017
Topic outline
-
-
Introduction
Proteins are the "worker" molecules of a cell. Their correct functioning and interaction with other molecules in the cell is crucial for many aspects of life. Proteins are involved in transport (hemoglobin: oxygen), storage (myoglobin in the muscle: oxygen), movement (dynein, actin), recognition (insulin receptors: protein kinase activation through binding of the hormone insulin), signal transduction (epidermal growth factor: cell growth), protection (antibodies), energy storage (glycogenin: glycogen), metabolism (F1-ATPase: ATP generation) and structure (keratin: skin).
The goal of this course is to revisit basic principles of protein structures and familiarize yourself with sources of experimental and theoretical protein structure models.
1 Backbone conformation and secondary structure
1.1 Peptide Torsion Angles
The figure below shows the three main chain torsion angles of a polypeptide. These are phi (Φ), psi (ψ), and omega (Ω).
The planarity of the peptide bond is restricted to 180 ° in nearly all of the main chain peptide bonds. In rare cases a deviation of 10 ° for a cis peptide bond is observed, that usually involves Proline.
1.2 The Ramachandran Plot
In a polypeptide the main chain N-Cα and Cα-C bonds are relatively free to rotate. These rotations are represented by the torsion angles phi (Φ: C-N-Cα-C) and psi (ψ: N-Cα-C-N), respectively.
G.N. Ramachandran used computer models of small polypeptides to systematically vary torsional angles with the objective of finding stable conformations. For each conformation, the structure was examined for close contacts between atoms. Atoms were treated as hard spheres with dimensions corresponding to their van der Waals radii. Angles, which cause spheres to collide correspond to sterically disallowed conformations of the polypeptide backbone.

In the diagram above the dark blue and black areas correspond to conformations where atoms in the polypeptide come closer than the sum of their van der Waals radii. These regions are sterically disallowed for all amino acids except Glycine, which is unique in that it lacks a side chain. The red/yellow regions correspond to conformations where there are no steric clashes, i.e. these are the allowed regions namely the α-helical and β-sheet conformations. The green areas show the allowed regions if slightly shorter van der Waals radii are used in the calculation, i.e. the atoms are allowed to come a little closer together. This brings out an additional region which corresponds to the left-handed α-helix.
L-amino acids cannot form extended regions of left-handed helix but occasionally individual residues adopt this conformation. These residues are usually Glycine but can also be Asparagine or Aspartate where the side chain forms a hydrogen bond with the main chain and therefore stabilizes this otherwise unfavourable conformation. The 310 helix occurs close to the upper right of the α-helical region and is on the edge of the allowed region indicating lower stability.
Disallowed regions generally involve steric hindrance between the side chain CH2 group and main chain atoms. Glycine has no side chain and therefore can adopt phi and psi angles in all four quadrants of the Ramachandran plot. Hence it frequently occurs in turn regions of proteins where any other residue would be sterically hindered.
Below is a Ramachandran plot of a protein containing almost exclusively β-strands (yellow dots) and only one helix (red dots). Note how few residues are out of the allowed regions. Note also that they are almost all Glycines (depicted with a little square instead of a cross).

Observe the effect of minor phi (Φ) and psi (ψ) angle changes:
1.2 The α-helix.
1.2.1 Development of an α-helix structure model.
Pauling and Corey twisted models of polypeptides around to find ways of getting the backbone into regular conformations which would agree with α-keratin fibre diffraction data. The most simple and elegant arrangement is a right-handed spiral conformation known as the 'α-helix'.
An easy way to remember how a right-handed helix differs from a left-handed one is to hold both your hands in front of you with your thumbs pointing up and your fingers curled towards you. For each hand the thumbs indicate the direction of translation and the fingers indicate the direction of rotation.
1.2.2 Properties of the α-helix.
The structure repeats itself every 5.4 Å along the helix axis, i.e. we say that the α-helix has a pitch of 5.4 Å. α-helices have 3.6 amino acid residues per turn, i.e. a helix 36 amino acids long would form 10 turns. The separation of residues along the helix axis is 5.4/3.6 or 1.5 Å, i.e. the α-helix has a rise per residue of 1.5 Å.
- Every main chain C=O and N-H group is hydrogen-bonded to a peptide bond 4 residues away (i.e. Oi to Ni+4). This gives a very regular, stable arrangement.
- The peptide planes are roughly parallel with the helix axis and the dipoles within the helix are aligned, i.e. all C=O groups point in the same direction and all N-H groups point the other way. Side chains point outward from helix axis and are generally oriented towards its amino-terminal end.

All the amino acids have negative phi and psi angles, typical values being -60 ° and -50 ° , respectively.
1.2.3 Distortions of α-helices.
The majority of α-helices in globular proteins are curved or distorted somewhat compared with the standard Pauling-Corey model. These distortions arise from several factors including:
- The packing of buried helices against other secondary structure elements in the core of the protein.
- Proline residues induce distortions of around 20 ° in the direction of the helix axis. This is because Proline cannot form a regular α-helix due to steric hindrance arising from its cyclic side chain which also blocks the main chain N atom and chemically prevents it forming a hydrogen bond. Proline causes two H-bonds in the helix to be broken since the NH group of the following residue is also prevented from forming a good hydrogen bond (read more). Helices containing Proline are usually long perhaps because shorter helices would be destabilized by the presence of a Proline residue too much. Proline occurs more commonly in extended regions of polypeptide.
- Solvent. Exposed helices are often bent away from the solvent region. This is because the exposed C=O groups tend to point towards solvent to maximize their H-bonding capacity, i.e. tend to form H-bonds to solvent as well as N-H groups. This gives rise to a bend in the helix axis.

310-Helices.
Strictly, these form a distinct class of helix but they are always short and frequently occur at the termini of regular α-helices. The name 310 arises because there are three residues per turn and ten atoms enclosed in a ring formed by each hydrogen bond (note the hydrogen atom is included in this count). There are main chain hydrogen bonds between residues separated by three residues along the chain (i.e. Oi to Ni+3). In this nomenclature the Pauling-Corey α-helix is a 3.613-helix. The dipoles of the 310-helix are not so well aligned as in the α-helix, i.e. it is a less stable structure and side chain packing is less favourable.

1.3 The β-sheet.
1.3.1 The β-sheet structure.
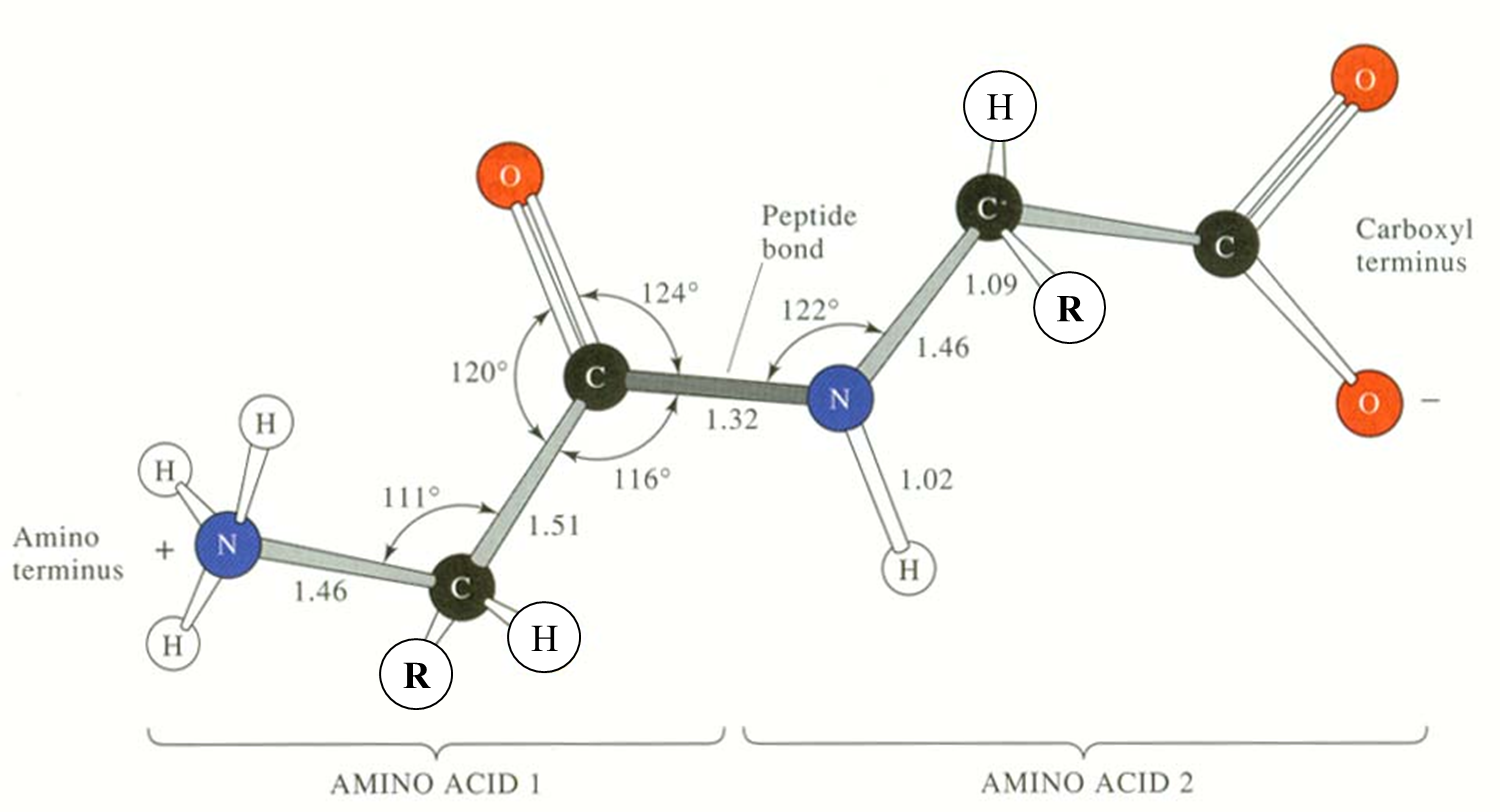
Pauling and Corey derived a model for the conformation of fibrous proteins known as β-keratins. In this conformation the polypeptide does not form a coil. Instead, it zig-zags in a more extended conformation than the α-helix. Amino acid residues in the β-conformation have negative Φ angles and the Ψ angles are positive. Typical values are Φ = -140 ° and Ψ = 130 ° . In contrast, α-helical residues have both Φ and Ψ negative. A section of polypeptide with residues in the β-conformation is referred to as a β-strand and these strands can associate by main chain hydrogen bonding interactions to form a sheet.
In a β-sheet two or more polypeptide chains run alongside each other and are linked in a regular manner by hydrogen bonds between the main chain C=O and N-H groups. Therefore all hydrogen bonds in a β-sheet are between different segments of polypeptide. This contrasts with the α-helix where all hydrogen bonds involve the same element of secondary structure. The R-groups (side chains) of neighboring residues in a β-strand point in opposite directions.

Imagining two strands parallel to this, one above the plane of the screen and one behind, it is possible to grasp how the pleated appearance of the β-sheet arises. Note that peptide groups of adjacent residues point in opposite directions whereas with a-helices the peptide bonds all point one way:
The axial distance between adjacent residues is 3.5 Å. There are two residues per repeat unit which gives the β-strand a 7 Å pitch. This compares with the α-helix where the axial distance between adjacent residues is only 1.5 Å. Clearly, polypeptides in the β-conformation are far more extended than those in the α-helical conformation.
1.3.2 Parallel, anti-parallel and mixed β-sheets.
In parallel β-sheets the strands all run in one direction, whereas in anti-parallel sheets they all run in opposite directions. In mixed sheets some strands are parallel and others are anti-parallel.
Below is a diagram of a three-stranded anti-parallel β-sheet. It emphasizes the highly regular pattern of hydrogen bonds between the main chain NH and CO groups of the constituent strands.
In the classical Pauling-Corey model the parallel β-sheet is somewhat more distorted and consequently exhibit weaker hydrogen bonds between the strands.
β-sheets are very common in globular proteins and most contain less than six strands. The width of a six-stranded β-sheet is approximately 25 Å. No preference for parallel or anti-parallel β-sheets is observed, but parallel sheets with less than four strands are rare, perhaps reflecting their lower stability. Sheets tend to be either all parallel or all anti-parallel, but mixed sheets do occur.

The Pauling-Corey model of the β-sheet is planar. However, most β-sheets found in globular protein X-ray structures are twisted. This twist is left-handed as shown below. The overall twisting of the sheet results from a relative rotation of each residue in the strands by 30 ° per amino acid in a right-handed sense.
Parallel sheets are less twisted than anti-parallel and are always buried. In contrast, anti-parallel sheets can withstand greater distortions (twisting and b-bulges) and greater exposure to solvent. This implies that anti-parallel sheets are more stable than parallel ones which is consistent both with the hydrogen bond geometry and the fact that small parallel sheets rarely occur (see above).

The left-handed twist of beta sheets amounts to 25º between each neighboring strands.
1.4 Reverse turns
A reverse turn is region of the polypeptide having a hydrogen bond from one main chain carbonyl oxygen to the main chain N-H group 3 residues along the chain (i.e. Oi to Ni+3). Helical regions are excluded from this definition and turns between β-strands form a special class of turn known as the β-hairpin (see later). Reverse turns are very abundant in globular proteins and generally occur at the surface of the molecule. It has been suggested that turn regions act as nucleation centres during protein folding.
Reverse turns are divided into classes based on the Φ and Ψ angles of the residues at positions i+1 and i+2. Types I and II shown in the figure below are the most common reverse turns, the essential difference between them being the orientation of the peptide bond between residues at (i+1) and (i+2).

The torsion angles for the residues (i+1) and (i+2) in the two types of turn lie in distinct regions of the Ramachandran plot.

Note that the (i+2) residue of the type II turn lies in a region of the Ramachandran plot which can only be occupied by Glycine. From the diagram of this turn it can be seen that were the (i+2) residue to have a side chain, there would be steric hindrance with the carbonyl oxygen of the preceding residue. Hence, the (i+2) residue of type II reverse turns is nearly always Glycine.
-
Protein structure information is represented in data formats specifying the coordinates and properties of individual atoms. A variety of different visualization and abstraction methods has been developed to allow a human eye to comprehend molecular properties of complex macromolecules.
Depending on which area you work in, certain preferences for representing molecular information are used interchangeably for Alanine:
A
Ala
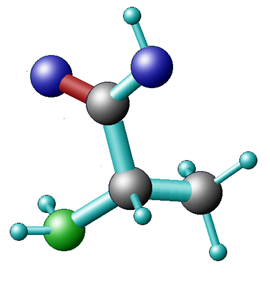
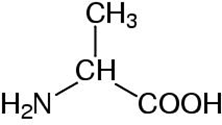
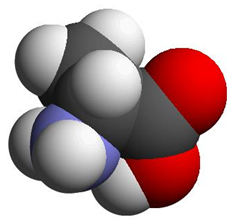
a potential computational chemists view chemical structure formula CPK representation The International Chemical Identifier (InChI) is used to unambiguously represent chemical entities in a string format:
InChI=1S/C3H7NO2/c1-2(4)3(5)6/h2H,4H2,1H3,(H,5,6)/t2-/m0/s1
The InChI notation is used for ligands, not for entire proteins.
Another very common notation is the widely used Simplified molecular-input line-entry system (SMILES) format (also for Alanine), although that is known to have short-comings concerning stereochemistry:
C[C@@H](C(=O)O)NThe SMILES notation is also used for ligands, not for entire proteins.
Likewise structural biologists have (ab-)used the ProteinDataBank (PDB) format intensively from the beginning. The format is column-based and thus ill-suited to describe entries with more than e.g. 99999 atoms or 9999 residues (see below for a superceding format PDBx/mmcif). There is also a the new mmtf format, which is beyond the scope of this course.
ATOM 263 N ALA A 35 1.429 34.959 -16.825 1.00 35.48 N
ATOM 264 CA ALA A 35 0.523 34.398 -17.829 1.00 35.10 C
ATOM 265 C ALA A 35 -0.724 33.878 -17.157 1.00 33.88 C
ATOM 266 O ALA A 35 -1.850 34.138 -17.600 1.00 33.13 O
ATOM 267 CB ALA A 35 1.209 33.268 -18.594 1.00 33.84 C
Recently the PDBX/mmcif format has been chosen as the main representation for all molecular data.
The format is easily extensible for more complex biological structures. The mmcif structure for Alanine:data_ALA # _chem_comp.id ALA _chem_comp.name ALANINE _chem_comp.type "L-PEPTIDE LINKING" _chem_comp.pdbx_type ATOMP _chem_comp.formula "C3 H7 N O2" _chem_comp.mon_nstd_parent_comp_id ? _chem_comp.pdbx_synonyms ? _chem_comp.pdbx_formal_charge 0 _chem_comp.pdbx_initial_date 1999-07-08 _chem_comp.pdbx_modified_date 2011-06-04 _chem_comp.pdbx_ambiguous_flag N _chem_comp.pdbx_release_status REL _chem_comp.pdbx_replaced_by ? _chem_comp.pdbx_replaces ? _chem_comp.formula_weight 89.093 _chem_comp.one_letter_code A _chem_comp.three_letter_code ALA _chem_comp.pdbx_model_coordinates_details ? _chem_comp.pdbx_model_coordinates_missing_flag N _chem_comp.pdbx_ideal_coordinates_details ? _chem_comp.pdbx_ideal_coordinates_missing_flag N _chem_comp.pdbx_model_coordinates_db_code ? _chem_comp.pdbx_subcomponent_list ? _chem_comp.pdbx_processing_site RCSB # loop_ _chem_comp_atom.comp_id _chem_comp_atom.atom_id _chem_comp_atom.alt_atom_id _chem_comp_atom.type_symbol _chem_comp_atom.charge _chem_comp_atom.pdbx_align _chem_comp_atom.pdbx_aromatic_flag _chem_comp_atom.pdbx_leaving_atom_flag _chem_comp_atom.pdbx_stereo_config _chem_comp_atom.model_Cartn_x _chem_comp_atom.model_Cartn_y _chem_comp_atom.model_Cartn_z _chem_comp_atom.pdbx_model_Cartn_x_ideal _chem_comp_atom.pdbx_model_Cartn_y_ideal _chem_comp_atom.pdbx_model_Cartn_z_ideal _chem_comp_atom.pdbx_component_atom_id _chem_comp_atom.pdbx_component_comp_id _chem_comp_atom.pdbx_ordinal ALA N N N 0 1 N N N 2.281 26.213 12.804 -0.966 0.493 1.500 N ALA 1 ALA CA CA C 0 1 N N S 1.169 26.942 13.411 0.257 0.418 0.692 CA ALA 2 ALA C C C 0 1 N N N 1.539 28.344 13.874 -0.094 0.017 -0.716 C ALA 3 ALA O O O 0 1 N N N 2.709 28.647 14.114 -1.056 -0.682 -0.923 O ALA 4 ALA CB CB C 0 1 N N N 0.601 26.143 14.574 1.204 -0.620 1.296 CB ALA 5 ALA OXT OXT O 0 1 N Y N 0.523 29.194 13.997 0.661 0.439 -1.742 OXT ALA 6 ALA H H H 0 1 N N N 2.033 25.273 12.493 -1.383 -0.425 1.482 H ALA 7 ALA H2 HN2 H 0 1 N Y N 3.080 26.184 13.436 -0.676 0.661 2.452 H2 ALA 8 ALA HA HA H 0 1 N N N 0.399 27.067 12.613 0.746 1.392 0.682 HA ALA 9 ALA HB1 1HB H 0 1 N N N -0.247 26.699 15.037 1.459 -0.330 2.316 HB1 ALA 10 ALA HB2 2HB H 0 1 N N N 0.308 25.110 14.270 0.715 -1.594 1.307 HB2 ALA 11 ALA HB3 3HB H 0 1 N N N 1.384 25.876 15.321 2.113 -0.676 0.697 HB3 ALA 12 ALA HXT HXT H 0 1 N Y N 0.753 30.069 14.286 0.435 0.182 -2.647 HXT ALA 13 # loop_ _chem_comp_bond.comp_id _chem_comp_bond.atom_id_1 _chem_comp_bond.atom_id_2 _chem_comp_bond.value_order _chem_comp_bond.pdbx_aromatic_flag _chem_comp_bond.pdbx_stereo_config _chem_comp_bond.pdbx_ordinal ALA N CA SING N N 1 ALA N H SING N N 2 ALA N H2 SING N N 3 ALA CA C SING N N 4 ALA CA CB SING N N 5 ALA CA HA SING N N 6 ALA C O DOUB N N 7 ALA C OXT SING N N 8 ALA CB HB1 SING N N 9 ALA CB HB2 SING N N 10 ALA CB HB3 SING N N 11 ALA OXT HXT SING N N 12 # loop_ _pdbx_chem_comp_descriptor.comp_id _pdbx_chem_comp_descriptor.type _pdbx_chem_comp_descriptor.program _pdbx_chem_comp_descriptor.program_version _pdbx_chem_comp_descriptor.descriptor ALA SMILES ACDLabs 10.04 "O=C(O)C(N)C" ALA SMILES_CANONICAL CACTVS 3.341 "C[C@H](N)C(O)=O" ALA SMILES CACTVS 3.341 "C[CH](N)C(O)=O" ALA SMILES_CANONICAL "OpenEye OEToolkits" 1.5.0 "C[C@@H](C(=O)O)N" ALA SMILES "OpenEye OEToolkits" 1.5.0 "CC(C(=O)O)N" ALA InChI InChI 1.03 "InChI=1S/C3H7NO2/c1-2(4)3(5)6/h2H,4H2,1H3,(H,5,6)/t2-/m0/s1" ALA InChIKey InChI 1.03 QNAYBMKLOCPYGJ-REOHCLBHSA-N # loop_ _pdbx_chem_comp_identifier.comp_id _pdbx_chem_comp_identifier.type _pdbx_chem_comp_identifier.program _pdbx_chem_comp_identifier.program_version _pdbx_chem_comp_identifier.identifier ALA "SYSTEMATIC NAME" ACDLabs 10.04 L-alanine ALA "SYSTEMATIC NAME" "OpenEye OEToolkits" 1.5.0 "(2S)-2-aminopropanoic acid" # loop_ _pdbx_chem_comp_audit.comp_id _pdbx_chem_comp_audit.action_type _pdbx_chem_comp_audit.date _pdbx_chem_comp_audit.processing_site ALA "Create component" 1999-07-08 RCSB ALA "Modify descriptor" 2011-06-04 RCSB #
-
In this chapter, we will explore different web interfaces for searching the archive of experimental protein structures and learn about their advantages.
The wwPDB is the single world-wide organization archiving experimental macromolecular structures. The archive currently holds over 100000 structures (updated number) and spans experimental data sources from Xray crystallography (XRAY), Nuclear Magnetic Resonance (NMR) or Electron Microscopy (EM). For a full list please see this link.
The rcsb.org web site is one of three main interfaces to the wwPDB archive. It features a very intuitive data presentation, that allows searching by simple text/keyword and features complex advanced queries as well. Please familiarize yourself with the summary page of 1AKE (please enter "1AKE" into the search field near the top of the screen).
Consider the panel describing experimental details of the deposition "1AKE", an adenylate kinase. Note that the resolution was determined to be 2.00 Å. Along with the space group the unit cell dimensions are given.
Now let us turn to the summary panel "molecular description". Please note that the details specified here consider the species the protein originated from along with gene names and if possible the protein's function. More detailed information is given in the graphical depiction "Protein Feature View".
The next panel of interest concerns structure validation. It is important to remind ourselves that checking the quality of any data we work with is crucial for determining its utility. Please inspect the Ramachandran plot for outliers.
Last let us inspect the ligand panel. Here the ligand with the three letter Code A5P is listed. Can you imagine why this ligand is present and why it is not possible to observe the natural substrate?
-
Several resources are providing easy access to protein structure information simply from a web interface. In this chapter, we will explore several commonly used web sites for exploring protein structures from a web browser.
The structure view of the PDB allows to experience various viewers. Please check them out!
The defacto standard JMOL
JMOL/JSMOL is the most common used web interface to molecular structure. <a href="https://www.stolaf.edu/people/hansonr/">Bob Hanson at St. Olaf</a> is the main developer. JMOL has a large community and many features, which makes it loved by chemists and structural biologists.
The Newcomers:
PV
PV is implemented in javascript rather than java, thereby alleviating the trouble connected with java plugins and ever-changing browser security settings. PV is fast and offers a pleasant style along with a powerful API to interface with it.
NGL
NGL is developed by Axel Rose and actively supported by the RCSB PDB. It features a very fast rendering and builds upon the new mmtf format for 3D structure information.
Proprietary:
MOLSOFT LLC develops ICMBrowser, which is available in a free version, some features require unlocking.
BioNext develops more than just a viewer, but it features great graphics! It is not free, though!
Others
Java based alternatives are Aquaria, OpenAstexViewer, both are not widely used, but Aquria has nice integrated data visualisation beyond structure and OpenAstexViewer appealing graphical defaults and powerful selections.
